
فقر الدم المنجلي
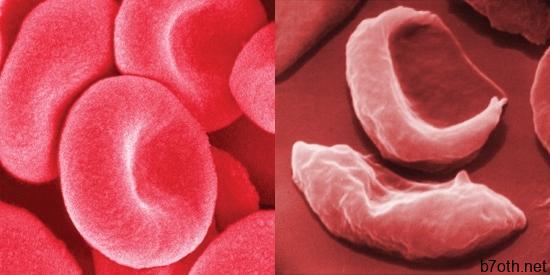
فقر الدم المنجلي ينجم عن نوع مختلف من الهيموغلوبين ، وهو البروتين الموجود فيه خلايا الدم الحمراء التي تحمل الأكسجين إلى أنسجة الجسم ، ودعا الهيموغلوبين S (HbS). HbS حساسة لنقص الأكسجين. عندما تقوم خلايا الدم الحمراء الحاملة بإفراج الأكسجين عن الأنسجة وتقليل تركيز الأكسجين داخل تلك الخلايا ، فإن HbS ، على النقيض من الهيموغلوبين الطبيعي (HbA) ، يصبح مكدسًا داخل الخلايا الحمراء في خيوط تتحول إلى قضبان حلزونية. ثم تتجمع هذه القضبان في حزم متوازية تشوه الخلايا وتطيلها ، مما يجعلها صلبة وتتخذ شكل المنجل. هذه الظاهرة قابلة للانعكاس إلى حد ما بعد أن تصبح الخلايا مؤكسجة مرة أخرى ، لكن المنجل المتكرر يؤدي في النهاية إلى تشويه لا رجعة فيه للخلايا الحمراء. تصبح الخلايا المنجلية مسدودة في الأوعية الدموية الصغيرة ، مما يتسبب في إعاقة الدورة الدموية الدقيقة ، مما يؤدي بدوره إلى تلف وتدمير الأنسجة المختلفة.
فقر الدم المنجلي ينجم عن وراثة جين الهيموغلوبين (HbS) المتغير من كلا الوالدين. (يُعرف هذا الميراث للجينات المتغيرة من كلا الوالدين بالحالة متماثلة الزيجوت). والشخص الذي يرث جين الخلية المنجلية من أحد الوالدين وجين الهيموغلوبين الطبيعي (HbA) من الوالد الآخر (الوراثة المعروفة باسم الحالة غير متجانسة الزيج) حامل لسمات الخلايا المنجلية. لأن خلايا الدم الحمراء للأشخاص غير المتجانسين تحتوي على كل من HbA و HbS ، فإن مثل هذه الخلايا تتطلب نزع الأكسجين بشكل أكبر بكثير لإنتاج منجل أكثر من تلك الموجودة في فقر الدم المنجلي. الغالبية العظمى من الأشخاص المصابين بسمة الخلايا المنجلية ليس لديهم أي أعراض للمرض ، على الرغم من ظهور بعض المظاهر – المرتبطة أساسًا بالمجهود القوي على ارتفاعات عالية -. لا يختلف معدل الوفيات الإجمالي للأشخاص المصابين بسمات الخلايا المنجلية عن معدل السكان العاديين المشابهين.
يحمل واحد من كل 12 من السود في جميع أنحاء العالم سمة الخلية المنجلية ، بينما يعاني واحد من كل 400 من المصابين بفقر الدم المنجلي. إذا كان كلا الوالدين يحملان سمة الخلية المنجلية ، فستكون فرص الإصابة 1 من كل 4 التي يصاب بها الطفل المولود لديه من فقر الدم المنجلي. ومع ذلك ، من خلال بزل السلى (تحليل السائل الأمنيوسي المحيط بالجنين) ، وهو إجراء اختبار يتم في المراحل المبكرة من الحمل ، من الممكن اكتشاف فقر الدم المنجلي في الجنين.
يتم توزيع جينات HbS جغرافيا في حزام استوائي واسع في أفريقيا ، كما توجد ، على الرغم من أنها أقل تواترا ، في أجزاء أخرى من القارة وفي الأمريكتين. تم تفسير استمرار HbS من خلال حقيقة أن الأشخاص غير المتجانسين يقاومون ملاريا. عندما يتم غزو الخلايا الحمراء للشخص المصاب بخاصية الخلية المنجلية بواسطة طفيل الملاريا ، تلتصق الخلايا الحمراء بجدران الأوعية الدموية ، وتتخلص من الأكسجين ، وتتخذ شكل المنجل ، ثم يتم تدميرها ، ويتم تدمير الطفيلي معها.
لا يوجد علاج لفقر الدم المنجلي. يكرس معظم الرعاية لتخفيف الأعراض. يتم إعطاء الرضع والأطفال الصغار المصابين بهذا المرض جرعات يومية منتظمة من البنسلين للوقاية من الإصابة الخطيرة. في بعض الحالات ، يتم نقل الدم بانتظام لمنع تلف الأعضاء والسكتة الدماغية وتخفيف أسوأ أعراض فقدان خلايا الدم الحمراء. في الحالات الشديدة كانت زراعة نخاع العظام ذات فائدة. المخدرات هيدروكسي يوريا يقلل من الأعراض الرئيسية لفقر الدم المنجلي. يبدو أن هيدروكسي يوريا ينشط الجين الذي يحفز إنتاج الجسم للهيموغلوبين الجنيني. هذا النوع من الهيموغلوبين ، الذي يتم إنتاجه عادة بكميات كبيرة فقط من قبل الأطفال قبل فترة وجيزة وبعد الولادة ، لا ينجل. يزيد علاج هيدروكسي يوريا من نسبة الهيموغلوبين الجنيني في مجرى الدم للمرضى البالغين من 1 إلى 20 في المائة ، وهي نسبة عالية بما يكفي لتقليل مشاكل الدورة الدموية التي تنشأ أثناء الأزمات بشكل ملحوظ.




